Genecensus: A hierachical system for comparative genomics study
The system provides the information of genomes at different levels: the relationship amongst different genomes, individual genomes and individual ORF in these genomes.
Tree Server: (Ref. [Lin])
Genecensus, as a database for comparative genomics, provides a resource for investigating different aspects of the genome and a reference point for analysis. One overview method of genome comparison is through the use of clusterings or phylogenetic trees based on different characteristics that relate the organisms.The GeneCensus Tree Server allows for a convenient online view for constructed trees and also acts as a tool for comparing trees to different standards. Simply, the architecture of the tree server is two-dimensional. One has the opportunity of choosing which tree they want to see, which are shown by tabs at the top of the screen, with a chose of different views that want to compare the tree to. This allows for all the trees to be analyzed from different angles.
For all the views, except the rRNA tree view, the first column in the view show buttons for Yale Resources. This provides links to specific research that has been done. This allows for connection to Yale reports on folds, on domains, or on genes. Some organisms have two links, because of extra genomes specific research that has been done. When coupled with all the reports on ORFs, this provides a powerful tool for analysis for levels as large as comparative genomics, to other levels as specific as individual ORF views.
Furthermore, each of the trees that are shown have a download feature above the tree that is shown. This allow for the easy download of the treefiles for each of the trees, with are in nexus format and can be easily viewed under PAUP, TreeView, or Phylip.
The trees that are currently available are subdivided to several sections represented by tabs on the top of the screen. Once a tab is selected, one can click on the trees available for that section. The tab that is currently selected is lit up. Currently, the tabs include ribosome, folds, sfams, COGs, composition, and ORFs.
- ribosome - this first tab show trees that are built based on the similarity of the ribosomal RNA. The traditional method for phylogenetic analysis is based on the small subunit ribosomal RNA (SSU rRNA), which is shown for both 8 and 20 genomes. For comparison, the trees based on the large subunit (LSU) for 8 genomes is also shown.
- folds - the second tab is a category of trees that are built based on the presence of absence of folds in different organisms. Furthermore, the occurrence of folds can be subdivided into the fold classes (all alpha, all beta, alpha + beta, and alpha / beta) and the data can be used for the construction of trees. A further comparison in this category shows the difference between the distance-based and parsimony techniques that were used for tree-building. The default method used for the comparison is distance-based.
- sfams - this third tab shows trees built on the occurrence of superfamilies in different organisms. Superfamilies less general structural groupings than folds and because of its greater number, have been found to be more differentiating, producing trees that are more similar to the traditional phylogeny. For this group, trees were built both for 8 and 20 genomes and also for both distance-based and parsimony methods.
- COGs - the fourth tab provides comparison of genomes based on the occurrence of homologous genes, also know as clusters of orthologous genes (COGs), which was obtained from the NCBI web site. All the COGs can be subdivided into three major groupings for comparison (Metabolism, Celluluar Processes, Information Storage and Processing). Furthermore, within these subjects, each class of orthologous genes can also be used for comparison, which are shown by the letters that stand for the class. Below in a table to describes these difference classes.
- composition - the fifth tab shows a category of trees that are built on the simple composition of the amino acids and di-nucleotides for 8 and 20 genomes. The trees that are marked raw are based on the absolute number of amino acids and di-nucleotides to generate a vector, and the distance calculated. For the other trees, the numbers calculated is normalized by the total number, producing percentages, which were used to generate a distance matrix for tree construction.
- ORFs - this final tab show trees that are built on the sequence similarity of homologous genes for the 8 genomes. Genes were chosen that were present in all the 8 genomes only once, in order that paralogous genes were not a factor. The number that are shown are the COG id numbers provided by NCBI.
- folds - the second tab is a category of trees that are built based on the presence of absence of folds in different organisms. Furthermore, the occurrence of folds can be subdivided into the fold classes (all alpha, all beta, alpha + beta, and alpha / beta) and the data can be used for the construction of trees. A further comparison in this category shows the difference between the distance-based and parsimony techniques that were used for tree-building. The default method used for the comparison is distance-based.
- taxonomy - this view provides a color coded listing of the accepted taxonomy as received from NCBI. Upon clicking each of the links, a small pop-up will give a small explanation of superkingdom, kingdom, phylum, or class. The superkingdom is represented as icons, a blue A for archaea, a red B for bacteria, and a green E for eukaryota. This allows for a quick view of how the tree under analysis corresponds to the accepted taxonomy. Each of the subsequent classes under the superkingdom is color coded to show similarity among the organisms. This is best seen with the ribosomal RNA tree for 20 genomes. The eukaryotes are colored green, darker for the metazoas and lighter for fungi. For the four archaeas that are marked blue, they are in the same kingdom of euryarchaeota. Each branching from that is given a different shade of blue. The majority of the organisms in under study are bacteria with the color red. Aquifex is the closest bacteria to archaea and thus has a purplish color. Each kingdom of the bacteria is given a separate shade of red, and differences within a kingdom is given off colors of that.
- composition - this second view shows some basic information for the composition of the whole genome, including genome size, number of genes, AT content and GC count. One specific view with the amino acid composition for 20 genomes in this particular view shows that the tree is corresponds closely to the AT content of the genome, while yet another view, clearly shows that without normalization, the raw di-nucleotide numbers is greatly skewed by the genome size and the number of genes.
- fold - the third view shows the number of folds within different fold classes, included all alpha, all beta, alpha + beta, alpha / beat, and multi folds. This allow for analysis of trees to see if these numbers provide any alternation of the trees. This effect, as expected, is most observed in the trees constructed on fold occurrence.
- superfamily - the fourth view displays the number for superfamily for each of the genomes within the superfamily classes, including all alpha, all beta, alpha + beta, alpha / beta, multi, and small. Like the folds, this analyzes the structural characteristics of the genome. As aforementioned, this sub-level of folds has more characteristics for distinguishing the genomes. Again, this view is perhaps most helpful in analyzing trees using the occurrence of superfamilies in the genomes.
- old COG - the fifth view shows the numbers of COGs for 8 genomes originally in the COGs database, for the total number and the larger subsets, including Metabolism, Cellular Processes, and Information Storage and Processing.
- new COG - the sixth view is analogous to the previous view. However, it incorporates the new data in the COGs database, which has 21 organisms. The same columns are shown.
- rRNA tree - this last view allows for a picture view of the tree under study side-by-side with the traditional ribosomal tree. This view automatically displays the ribosomal tree with 8 and 20 genomes, depending on how many of genomes are in the tree under review. This allows for a more intuitive comparison, and when coupled with the first taxonomy view, provides a powerful tool to compare the tree with the established phylogeny.
- composition - this second view shows some basic information for the composition of the whole genome, including genome size, number of genes, AT content and GC count. One specific view with the amino acid composition for 20 genomes in this particular view shows that the tree is corresponds closely to the AT content of the genome, while yet another view, clearly shows that without normalization, the raw di-nucleotide numbers is greatly skewed by the genome size and the number of genes.
With the two dimensions combined with existing trees, the tree server allows for easy perusal of ready built trees in comparison of specific features. With combined with more detailed organisms pages, and ORF reports, this resource allow for the exploration of the whole genomes on many different levels.
J Translation, ribosomal structure & biogenesis
K Transcription
L DNA replication, recombination & repair
D Cell division and chromosome partitioning turnover, chaperones
O Posttranslational modification, protein
M Cell envelope biogenesis, outer membrane
N Cell motility & secretion
P Inorganic ion transport & metabolism
T Signal transduction mechanisms
C Energy production & conversion
G Carbohydrate transport & metabolism
E Amino acid transport & metabolism
F Nucleotide transport & metabolism
H Coenzyme metabolism
I Lipid metabolism
Genome Pages:
Genome Pages provide the information for the first 20 completely sequenced genomes, including 4 archaea, 2 eukayotes and 16 bacteria. A search box is shown for user to search the individual ORF. For detail of this search tool, See smartlink. The work from this group includes: statistical analysis of thermophilic and mesophilic genomes, pair composition analysis of transmembrane sequence in these genomes. Users can also download the tables of these results. External links concerning these genomes are also presented. In addition, the pages are designed to be open system that users can link their related work from these pages.
SmartLink:
We provide a search facility called smartlink. It allows users to enter a ORF name and obtain the information concerning the ORF, which mainly inlcudes the structural annotation (results from the group) and other resources. SmartLink has the following features:
- it supports several ORF identifier system. User can either input the ORF name (e.g. MG001), SwissProt Name (e.g. DP3B_MYCGE), SwissProt Accession Number (e.g. P47247), genebank PID (e.g. 3844620), Protein Accession Number (e.g. AAC71217.1), or gene name (e.g. dnaN).
- it provides other resources concerning the ORF. The other resources include SwissProt Niceprot, NCBI annotation, Tigr annotation, and GenProEC.
- it provides information on different level of detail: for yeast, worm, and Mycoplasma genitalium, special pages were made. These pages include more information. For other ORFs which were studied in the group, a graphical structural annotation (ORF report) is presented with other resources (see 2). If the ORF hasn't been studied in the group, simply other resources are given.
- graphical presentation of the structural annotations: orfreport
orfreport: (Ref. [Gerstein97][Gerstein98])
A plot is presented for the structural annotations, including PDB match, low complexity (a region of sequence with highly biased composition), transmembrane, signal sequence (a local functional site), linker, uncharacterized region, all alpha, all beta regions, TM best, PDB match via PSI-Blast.
PDB match and PSI-Blast match: a structure was assigned to a segment of sequence if the segments were found to be homologous to a known structure in PDB. The methods used are standard Blast, FASTA or PSI-Blast.
Low complexity: stretch of low complexity sequence are thought not to fold into globular protein structures, they may correspond to fibrous or disordered structures. They were identified with SEG program using standard parameters K(1)=3.4, K(2)=3.75, and a window size of 45.
Transmembrane or TM best: Transmembrane segments were identified using the GES hydrophobicity scales. The values from the scale for amino acids in a window of size 20 (the typical size of a transmembrane helix) were averaged and then compared against a cutoff of -1kcal/mol.
Linker: short sequences between characterized segments (PDB matches, low complexity regions and transmembrane helices) are considered as linkers.
Uncharacterized regions: The sequences which are neither characterized segments nor linkers are called uncharacterized regions.
Secondary structures: program GOR was used to predict the secondary structures.
Mouseover can show information (position, fold name, match score, etc.) about the segment.
Below the plot, segment sequences are shown. For the segments larger than 30, a link to Blast search for this segment is given. For the transmembrane segment, a pair pattern analysis (see ...) is given.
Statistical analysis of amino acid pairs in transmembrane sequences (Ref.[Senes])
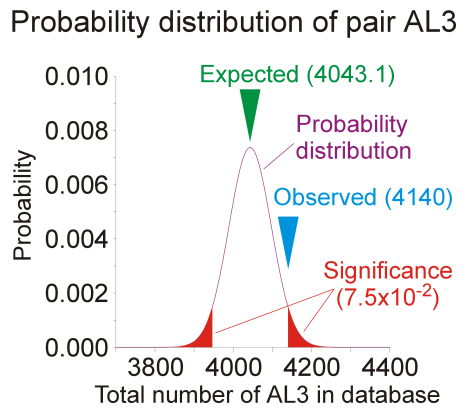
PairAny combination of an amino acid residue with another residue at separation i, i+k (k is called the register).
Example: AL3 correspond to a alanine residue and a leucine residue at i, i+3 (AxxL).
Observed
Observed total number of a particular pair found in a database of putative transmembrane helices. The database was obtained from all transmembrane domains annotated in Swiss-Prot (rel. 37) after homology removal.
Expected
Expected total number of the pair in the same database. The number is calculated as the average of a probability distribution calculated on any possible internal permutations of all sequences of the database.
St.Dev.
Standard deviation of the probability distribution.
Significance
Significance of the difference between the observed and expectated number of the pair, corresponding to the probability of observing an equal or larger difference if the residues were randomly distributed inside the sequences (p value). The value is obtained by integrating the probability distribution from the observed count to infinity and simmetrically on the other side.
Given the same odds ratio, the p value gets smaller increasing size of the database. For databases of very different size (as for the bitopic vs polytopic databases) p values cannot be compared
Odds ratio
Ratio between the observed and the expected occurrences of the pair. A ratio above one indicates that the pair is over-represented in the database with respect to its average expectation. A ratio below one indicates an under-represented pair.
Database
Databases of non-homologous transmembrane domains (TMs) obtained from all 46,946 TRANSMEM annotations in Swiss-Prot rel. 37, after homology removal, and from 20 genome databases.
Reference:
[Lin]: Lin J. and Gerstein, M. Whole-genome trees based on the occurrence of folds and orthologs: implications for comparing genomes on different levels. Genome Res. 2000 Jun;10(6):808-18. [HTML][DeRijk]: De Rijk, P., Robbrecht, E., de Hoog, S., Caers, A., Van de Peer, Y. & De Wachter, R. 1999. Database on the structure of large subunit ribosomal RNA. Nucleic Acids Res 27: 174--8. Doolittle, R. F. 1998. Microbial genomes opened up. Nature 392: 339--42.
[Hegyi]: Hegyi, H. & Gerstein, M. 1999. The relationship between protein structure and function: a comprehensive survey with application to the yeast genome. J Mol Biol 288: 147--64 [Medline]
[Felsenstein]: Felsenstein, J. 1993. PHYLIP (Phylogeny Inference Package) version 3.5c. Distributed by the author. Department of Genetics, University of Washington, Seattle.
[Gerstein97]: Gerstein, M. 1997. A Structural Census of Genomes: Comparing Eukaryotic, Bacterial and Archaeal Genomes in terms of Protein Structure. J. Mol. Biol. 274: 562--576. [Medline]
[Gerstein98]: Gerstein, M. 1998. Patterns of protein-fold usage in eight microbial genomes: a comprehensive structural census. Proteins 33: 518--34. [Medline]
[Swofford]: Swofford, D. L. 1998. PAUP*. Phylogenetic Analysis Using Parsimony (*and
Other Methods). Version 4. Sinauer Associates, Sunderland, Massachusetts.
[Woese]: Woese, C. R., Kandler, O. & Wheelis, M. L. 1990. Towards a natural system
of organisms:
proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad
Sci U S A 87:
4576--9.[Medline]
[Senes]: A. Senes, M. Gerstein and D.M. Engelman:
"Statistical analysis of amino acid patterns in transmembrane helices: the
GxxxG
motif occurs frequently and in association with beta-branched residues at
neighboring
positions."
J.Mol.Biol (2000), 296(3), 921-936 [Medline]