PartsList: a web-based system for dynamically ranking protein folds based on disparate attributes, including whole-genome expression and interaction information
Jiang Qian, Brad Stenger, Cyrus A. Wilson, Jimmy Lin, Ronald Jansen, Sarah Teichmann1, Jong Park2, Werner Krebs, Vadim Alexandrov, Nathaniel Echols, Mark Gerstein*Department of Molecular Biophysics and Biochemistry Yale University PO Box 208114, New Haven, CT 06520, USA
1Department Biochemistry & Molecular Biology, University College London, Darwin Bldg, Gower St, London WC1E 6BT, UK and 2European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, CB10 1SD, UK
*To whom correspondence should be addressed. Tel: +1 203 432 6105; Fax: +1 360 838 7861; Email: Mark.Gerstein@yale.edu
Abstract
As the number of protein folds is quite limited, a mode of analysis that will be increasingly common in the future, especially with the advent of structural genomics, is to survey and re-survey the finite parts list of folds from an expanding number of perspectives. We have developed a new resource, called PartsList, that lets one dynamically perform these comparative fold surveys. It is available on the web at http://bioinfo.mbb.yale.edu/partslist . The system is based on the existing fold classifications and functions as a form of companion annotation for them, providing "global views" of many already completed fold surveys. The central idea in the system is that of comparison through ranking; PartsList will rank the ~420 folds based on more than 180 attributes. These include: (i) occurrence in a number of completely sequenced genomes (e.g. it will show the most common folds in the worm vs yeast); (ii) occurrence in the structure databank (e.g. most common folds in the PDB); (iii) both absolute and relative gene expression information (e.g most changing folds in expression over the cell cycle); (iv) protein-protein interactions, based on yeast two-hybrid information and comprehensive PDB surveys (e.g. most interacting fold); (v) sensitivity to inserted transposons; (vi) the number of functions associated with the fold (e.g. most multi-functional folds); (vii) amino acid composition (e.g. most Cys-rich folds); (viii) protein motions (e.g. most mobile folds); and (ix) the level of similarity based on a comprehensive set of structural alignments (e.g. most structurally variable folds). The integration of whole-genome expression and protein-protein interaction data with structural information is a particularly novel feature of our system. We provide three ways of visualizing the rankings: a profiler emphasizing the progression of high and low ranks across many pre-selected attributes, a dynamic comparer for custom comparisons, and a numerical rankings correlator. These allow one to directly compare very different attributes of a fold (e.g. expression level and maximum motion) in the uniform numerical format of ranks. We also provide a traditional single-structure report to summarize much information related to genome occurrence, expression level, motion, function, and interaction in a single compact view with further links to many detailed resources.
Introduction
Protein folds can be considered as most basic molecular parts. There are a very limited number of them in biology. Currently, about 500 are known and it is believed that there may be no more than a few thousand in total (1-3). This number is considerably less than the number of genomes in complex, multicellular organisms (>10,000 for multicellular organisms (4)). Consequently, folds provide a valuable way of making manageable and simplifying complex genomic information. In addition, folds are useful for studying the relationships between evolutionarily distant organisms since in making comparisons structure is more conserved than sequence or function.
In a general sense, how should one approach the analysis of molecular parts? A simple analogy to mechanical parts may be useful in this regard. Given the ?parts? from a number of devices (e.g. a car, a bicycle, and a plane) one might like to know what ones are shared by all and which are unique (say wings for a plane). Furthermore, one might want to know which are common, generic parts and which are more specialized. Finally, one might like to organize the parts by a number of standardized attributes (e.g. the most flexible parts, the parts with the most functions, and the biggest parts). PartsList aims to provide answers to simple questions such as these for the domain of protein folds.
Properties related to protein folds can be divided into those that are "intrinsic" versus "extrinsic". Intrinsic information concerns an individual fold itself -- e.g. its sequence, 3D structure, and function -- while ?extrinsic? information relates to a fold in the context of all other folds -- e.g. its occurrence in many genomes and expression level in relation to that for other folds. Web-based search tools already provide intrinsic information about protein structures in the form of reports about individual structures. Valuable examples include the PDB Structure Explorer (5), PDBsum (6), and the MMDB (7). However, current resources lack the ability to fully present extrinsic information.
Likewise, while there are many databases storing information related to individual organisms (e.g. SGD, MIPS and FlyBase (8-10)), comparative genomics (PEDANT and COGs (9,11)), gene expression (GEO, the Gene Expression Omnibus at the NCBI, and ExpressDB (12)), and protein-protein interactions (DIP and BIND (13,14)), none of them integrates gene sequences, protein interactions, expression levels and other attributes with structure. (However, it should be mentioned the Sacc3D module of SGD and PEDANT do tabulate the occurrence of folds in genomes.)
PartsList is arranged somewhat differently from most other biological resources. In a usual database (e.g. GenBank(15)) the number of entries increases as the database develops, while each entry has a fairly fixed number of attributes to describe it. In contrast, PartsList is envisioned to have a relatively stable number of entries, i.e. the finite list of protein folds, while the attributes that describe each entry are expected to increase considerably. In the current version of PartsList the properties for a protein fold include: amino acid composition, alignment information, fold occurrences in various genomes, statistics related to motions, absolute expression levels of yeast in different experiments, relative expression ratios for yeast, worm, and E. coli in various conditions, information on protein-protein interactions (based on whole genome yeast interaction data and databank surveys), and sensitivity of the genes associated with the fold to inserted transposons.
One motivation to build the database is to compare protein folds in a rich context and in a unified way. This was achieved through ranking. This allows users to directly compare very different attributes of a fold in a uniform numerical format. The rankings can be visualized in three ways: a profiler emphasizing the progression of high and low ranks across many pre-selected attributes, a rankings comparer for custom comparisons, and a numerical rankings correlator. This can help users gain insight into the functions of protein folds in the context of the whole genome. Our system makes it very easy to answer questions like: "What is the most common fold in the worm as compared to E. coli?", "What is the most highly expressed fold in yeast and how does this compare to the fold that changes most in expression level during the cell-cycle?" And "which fold has the most protein-protein interactions in the PDB and is it highly ranked in terms of protein motions?"
PartsList is built on top of the Structural Classification of Protein (SCOP) (16) fold classification and acts as an accompanying annotation to this system. SCOP is divided into a hierarchy of five levels: class, fold, superfamily, family and protein. The "parts" in our system can be either SCOP folds or superfamilies. However, sometimes for ease of expression we will just refer to "folds" when really mean "folds and/or superfamiles." We currently use 420 folds and 610 superfamilies from in PartsList. Each is represented by a representative domain, which is also the key for each entry of protein fold.
While we chose to use the SCOP classification, we could equally well have based the system on the other existing fold classifications, e.g. CATH (17), FSSP (18), or VAST (19,20). Moreover, for most attributes, we could also have developed our system around non-structural classifications of protein parts -- e.g. Pfam (21), Blocks (22), or SMART (23). However, basing it around actual structural folds has the advantage that each part is more precisely and physically defined.
Attributes that can be ranked: Information in the system
Currently the attributes for each entry (i.e. protein fold) can be separated into several main categories: statistical information from a comprehensive set of structural alignments, amino-acid composition information, fold occurrences in various genomes, expression levels in different experiments, protein interactions, macromolecular motion, transposon sensitivity and miscellaneous.
We have developed a formalism for expressing each of the attributes, which is described in Table 1. In the table the term PART refers to either fold or superfamily, depending on which of these is being ranked. Essentially, we have a database of attributes where each attribute is given a standardized description and associated with a precise reference. In the following, we describe some main categories of attributes.
Genome Occurrence
The data in this category reveal fold occurrences in 20 different genomes, including 4 archaea, 2 eukaryotes, 16 bacteria; (additional details online).
The data were obtained in the following fashion: Once a library of folds has been constructed, representative sequences can be extracted (44). Then one can use these to search genomes by comparing each representative sequence against the genomes using the standard pairwise comparison programs, FASTA (49) and BLAST (50) and well-established thresholds (51).
Alternatively, one can build up profiles by running each representative sequence against PDB with PSI-Blast and then comparing these profiles against each of the genomes. This later procedure is more sensitive than pairwise comparison and relatively efficient once the profiles are made up. However, in doing large-scale surveys one has to be conscious of the potential biases introduced by the profiles being more sensitive for larger families, often resulting in the big families getting even bigger.
After the structure assignment, it becomes easy to enumerate how often a fold or structure feature occurs in a given genome or organism. Detailed information can be found in (24-26,52). This pools assignments are from previous work (53,54).
Alignment
Number Structures. We did a comprehensive set of structural alignments of structures in the PDB structure databank (29,55,56). The number of structures and aligned pairs used in these comparisons, which are based around Astral (44), give approximate measures of the occurrence of folds in the PDB. Comparison of these values to those for genome occurrence provides a measure of how biased the composition of the PDB is (57).
Sequence Diversity. The scores from the alignments indicate the sequence diversity between the related structures within folds or superfamilies, in terms of percent sequence identity and a sequence-based P-value. P-values are useful measures of statistical significance of the similarity calculation. A P-value is the probability that one can obtain the same or better alignment score from a randomly composed alignment. A smaller P-value is less likely to have been obtained by chance than a larger P-value. Large P-values close to 1.0 indicate that the similarity is characteristically random and thus insignificant.
Structural Diversity. We also give analogous measures of the diversity of the structures with fold, allowing one to rank folds by their degree of variability. We tabulate untrimmed and trimmed RMS, along with the structural P-value. RMS, root-mean-squared deviation in alpha carbon positions, has been the traditional statistic that gauges the divergence between two related structures. Smaller RMS scores indicate more closely related structures. However, sometimes a few ill-fitting atoms may significantly increase the RMS of structures known to be similar. To compensate for this we also report a "trimmed" RMS for a conserved core structure, which is based on the better fitting half of the aligned alpha-carbons, and structural P-value, which compensates for other effects such as structure size. For details, see Wilson et al. (29).
Composition
This allows us to see which folds are most biased in composition of particular amino acids. We use various levels of the Astral clustering of the SCOP sequences to arrive at the composition (44).
Expression
Three techniques are frequently used to obtain genome-wide gene expression data. They are Affymetrix oligonucleotide gene chips, SAGE (Serial Analysis of Gene Expression), and cDNA microarrays (37,58,59). SAGE and, to some degree, gene chips measure the absolute expression levels (in units of mRNA transcripts per cell), while microarrays are used to obtain the expression level changes of a given ORF as ratio to a reference state.
A main motivation for expression experiments often is to study protein function and to characterize the functions of unannotated genes. However, this does not preclude relating other attributes of proteins, such as their structure, to expression data. For instance, it may be that highly expressed protein folds share a number of characteristics, such a particularly stable architecture or a composition biased in certain way. Relating expression and structure involved matching the PDB structure database against the genome and then summing the expression levels of all ORFs containing the same fold.
Absolute. The absolute expression level data gives a good representation of highly expressed genes. All the experiments currently indexed by PartsList are for yeast. For each experiment, in addition to ranking based on the average expression level for a fold, we also consider the composition in the transcriptome and the enrichment of this value relative to its composition in the genome. Transcriptome composition is the fractional composition of a fold (relative to that for other folds) in the mRNA population. In other words, it is the composition of a fold in the genome weighted by the expression levels of each of the genes. The enrichment is the relative change between the composition of a fold in the genome and the transcriptome. For more details, see (27,60). We report values for experiments from a number of different labs (35-38) and a single reference set that merges and scales all the expression sets together.
Ratio. The expression ratio data shows the most actively changing genes over a period of time (e.g. cell cycle) or based on a change in states (e.g. healthy vs. diseased). Source data for expression ratios are the fluctuations in expression of a certain fold over a period of time (e.g. the cell cycle). These are measured in terms of standard deviations for a particular fold, which is calculated from the average of the expression ratio standard deviations for each gene that matches the fold structure.
Interactions
Information on protein-protein interactions is derived from surveys of the contacts in the PDB and the whole-genome experiments done on yeast.
PDB. To determine which domains interact with one another in the PDB entries indexed by SCOP (9,580 at the time of the analysis), the coordinates of each domain were parsed to check whether there are five or more contacts within 5 E to another domain. The distance of 5 E was chosen, as this is a conservative threshold for interaction between two atoms, where the atoms are either Ca?s or atoms in side-chains. The 5-contact threshold was chosen to make sure the contact between the domains was reasonably extensive. (In fact, the number of domains identified as contacting each other hardly changed for thresholds between 1 and 10 contacts and 3 to 6 E distances). The program for parsing PDB coordinates is available at ftp://ftp.ebi.ac.uk/pub/contrib/jong/PSIPFI/.
Yeast. The interactions between structural domains in the yeast genome were obtained by assigning protein structures to the yeast proteins as described above. Once these had been made, deriving a set of intramolecular interactions was straightforward. Structural domains contained within the same ORF that were within 30 amino acids were assumed to interact. (This is generally true of the domains in the PDB, with a few exceptions, such as domains in transcription factors like adjacent zinc fingers, or variable and constant immunoglobulin domains.) To derive intermolecular interactions in the yeast genome we combined three sets of protein-protein interactions: (i) the MIPS web pages on complexes and pairwise interactions (February 2000)(9), (ii) the global yeast-two-hybrid experiments by Uetz et al. (45) and (iii) large-scale yeast two-hybrid experiments by Ito et al. (46). Out of all these pairwise interactions known for yeast ORFs, there is a limited set in which both partners are completely covered by one structural domain (to within 100 residues). This set of protein pairs was used to derive a further set of domain contacts in the yeast genome (61).
Motions
Information on motions is from the Macromolecular Motions Database (30,31). We consider a set of approximately 4400 motions automatically identified by examining the PDB and a smaller, manually curated set of motions. For each fold we determine the number of entries in the motions database that are associated with it. Then over this set of motions we either average or take the maximum value of a number of relevant statistics describing the motion, i.e. the maximum Ca displacement in the motion, the overall rotation of the motion, the energy difference between the start and endpoints of structures involved in the motion.
Transposon Sensitivity
Ross-MacDonald et al. (34) developed a procedure for randomly inserting transposons throughout the yeast genome. They investigated the phenotypes resulting from each insertion in 20 different growth conditions in comparison to wild-type growth. The experiment for each insertion in each condition was repeated for several times. If the observed phenotype of the mutant deviates from the average wild-type phenotype, this could be either because of a real effect of the mutation on the cell or it could just a typical variation of the phenotype of wild-type cells. We developed a P-value score that measures the degree of confidence that the observed phenotype results from randomly changing wild-type cells. The negative logarithm of this P-value rises with the significance of the phenotype measurements and can be understood as the sensitivity of the cell to mutations in a particular gene. We calculated a value for the transposon sensitivity for protein folds by geometrically averaging the P-values of the associated genes.
Miscellaneous
The miscellaneous section includes any information that does not fit into a major category. It includes: number of pseudogenes in worm associated with a fold (47), total number of functions and number of enzymatic functions associated with a fold (48), the average length of the sequence, and the year the domain structure was originally determined.
Ranking all the folds based on extrinsic information
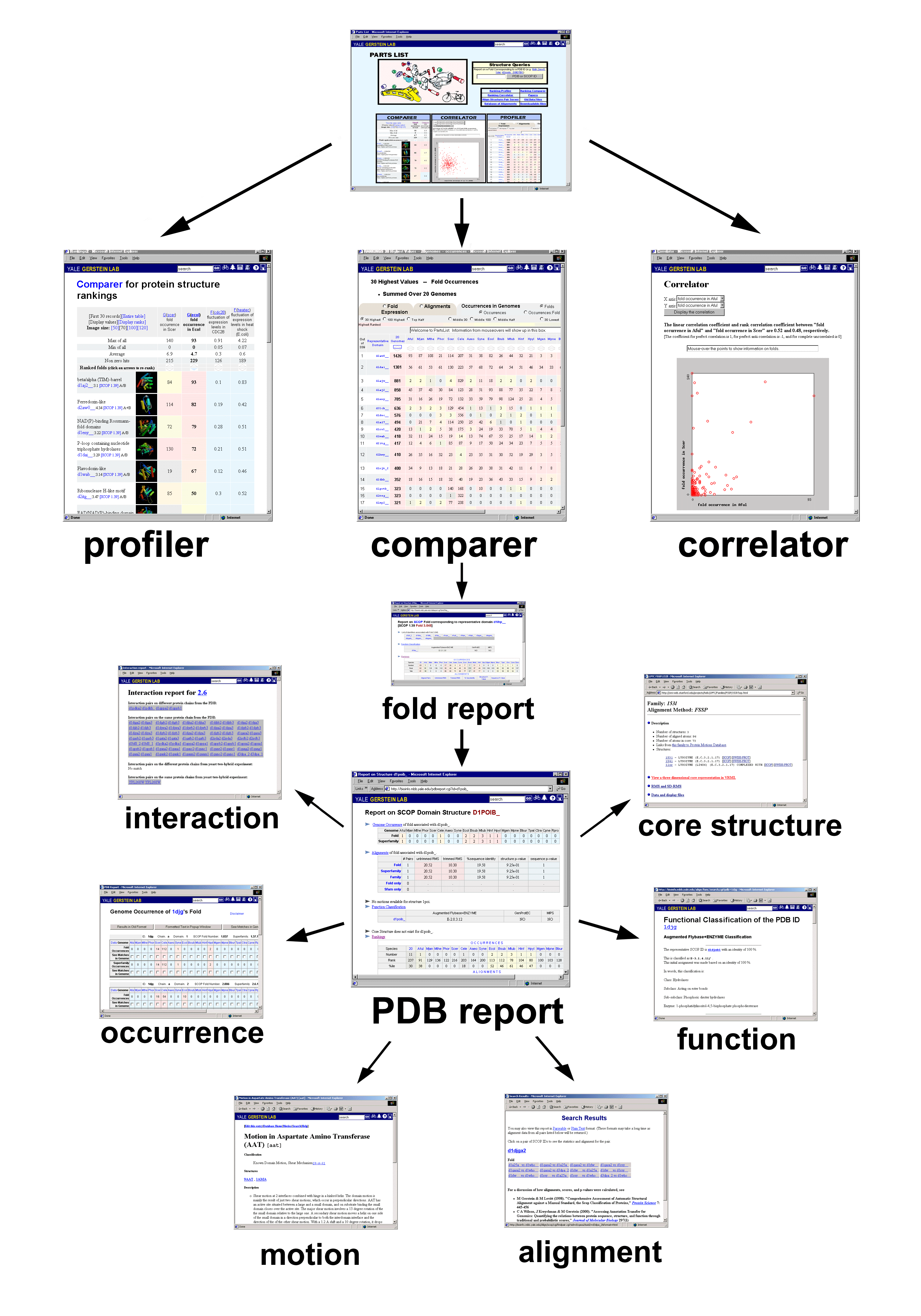
The PartsList resource facilitates exploring extrinsic information by dynamically ranking protein folds in different contexts, such as genome and expression levels. We provide three tools for visualizing the rankings: Comparer, Correlator, and Profiler. The overall structure of PartsList is schematically shown in Fig. 1. Note that the precise values for the rankings are contingent on the evolving contents of various databanks. Thus, over time as more structures are determined, one should expect statistics such as the most common folds in a particular genome to change somewhat.
Comparer
The motivation behind Comparer is to allow one to rank folds according to a given attribute and then see the ranks associated with other attributes. The ranking attribute and the additional attributes are selected by the user. Figure 2(a) shows an example. The most common folds in E. coli are shown alongside three other attributes: fold occurrence in yeast, fluctuation in expression level during the yeast cell cycle, and fluctuation in expression level in E. coli during heat shock. Which displayed attribute is used to rank the folds can be easily changed; in the example in Figure 2(a) the report can be re-sorted based on the other three attributes by clicking on arrows.
Profiler
In principle, Profiler presents the same information as Comparer. However, Profiler shows the progressing pattern for several pre-selected categories. Figure 2(b) shows an example that highlights the phylogenetic pattern of fold occurrence in 20 genomes.Correlator
Correlator uses linear and rank correlation coefficients to measure the association between two selected attributes. The difference between these two types of correlation coefficients is that the former relates to the actual values while the latter relates to the ranks among the samples. The interpretation of the linear correlation coefficient can be completely meaningless if the joint probability distribution of the variables is too different from a binormal distribution. This is the reason for introducing the rank correlation coefficient. Correlator provides both coefficients for the selected quantities. In most cases, they are close. For example, the linear correlation coefficient and rank correlation coefficient for fold occurrence in genomes A. fulgidus and M. jannaschii (Aful and Mjan) are 0.88 and 0.77, respectively, while the corresponding coefficients for fold occurrence in A. fulgidus and S. cerevisiae (Scer) are 0.52 and 0.48, respectively. This is not surprising because the first two genomes are both Archaeal, while in the second comparison one genome belongs to Archaea (Aful) and another to Eucarya (Scer). As one would expect, the fold occurrences for the more closely related genomes have a higher correlation.In addition to the coefficients, Correlator displays a scatter plot to aid in visualizing the correlation between the selected fold attributes. Figure 2(c) shows the scatter plot for the second example above: the correlation between occurrences in the A. fulgidus and S. cerevisiae genomes. One can easily observe that some folds appear frequently in Scer but seldom or never in A. fulgidus. By clicking on a point on the plot, one obtains detailed information about the corresponding fold. This kind of plot can reveal interesting folds with certain relationships between attributes even though in some cases the overall correlation coefficients between the two attributes are almost zero (i.e. no correlation).
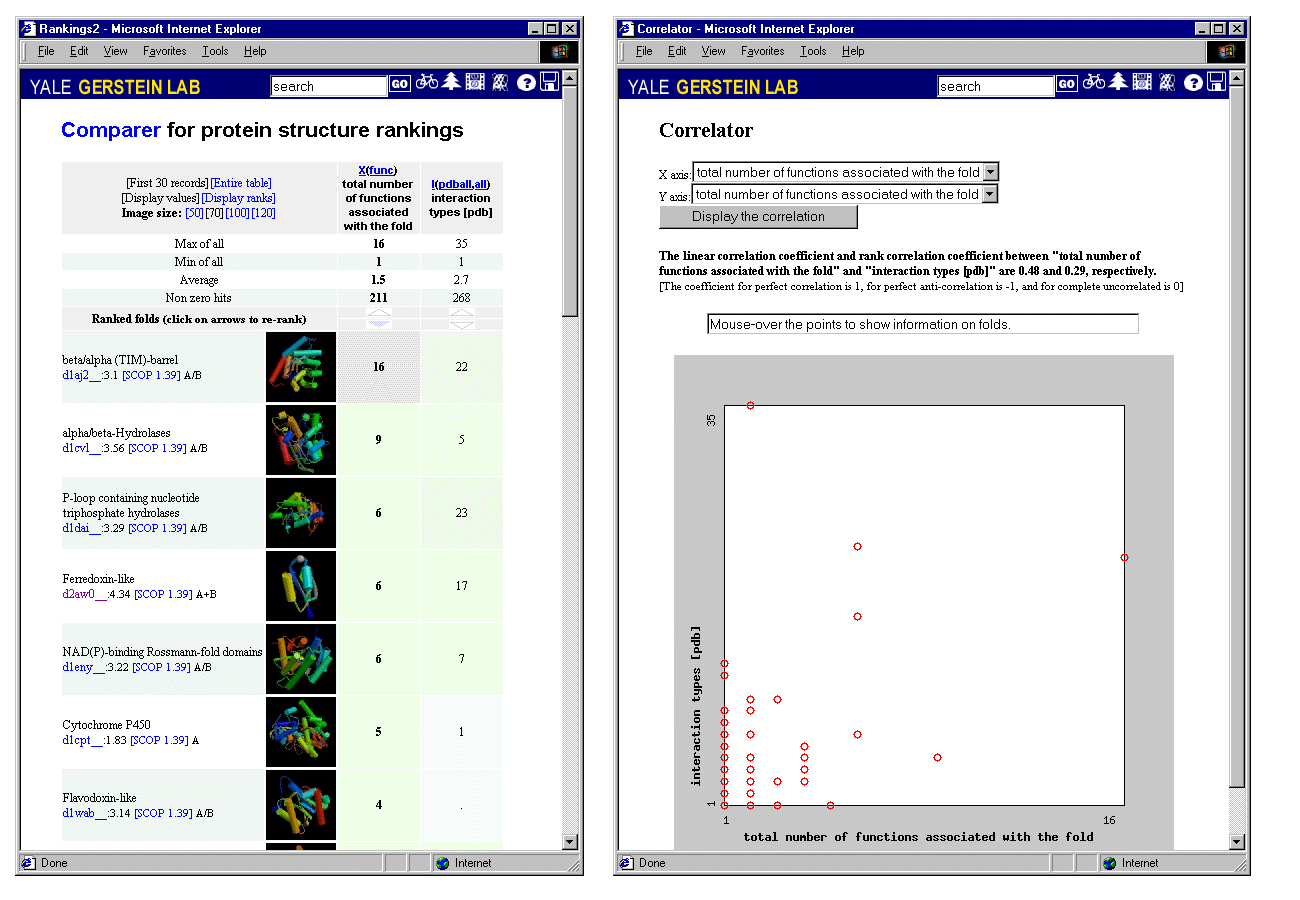
Going back and forth between Correlator and Comparer allows one to see interesting relationships between disparate attributes of proteins. Figure 3 illustrates a comparison of functions and interactions. It shows a ranking of the folds that have the most interactions in the PDB in comparison to the folds that have the most functions. It is immediately apparent that the most multi-functional folds also have the most distinct interactions with other folds.
Traditional Single-Structure reports
In addition to the tools that compare and relate the extrinsic properties of protein folds, we provide traditional reports that are more focused on an individual structure.
Occurrence report. This allows users to see the number of times that a fold corresponding to the queried protein structure occurs in various genomes. This gives a phylogenetic profile of the occurrence of a particular fold in 20 genomes, similar in spirit to the fold patterns discussed earlier (25).
Function report. This summarizes the functional classification of the queried PDB structure. It merges a number of functional classifications, including FlyBase(10), ENZYME(62), GenProEC(63) and MIPS(9).
Alignment report. This gives detailed information on structural alignments available between pairs of protein domains associated with a fold. A pair viewer is provided, which gives many key statistics about the alignment (e.g. RMS, sequence identity, number of fit atoms, etc.), in addition to a listing of the actual aligned residues. Both HTML and parseable text views are available.
Interaction report. This shows all the pairs of protein-protein interactions associated with a fold based on either the PDB survey or yeast genome data.
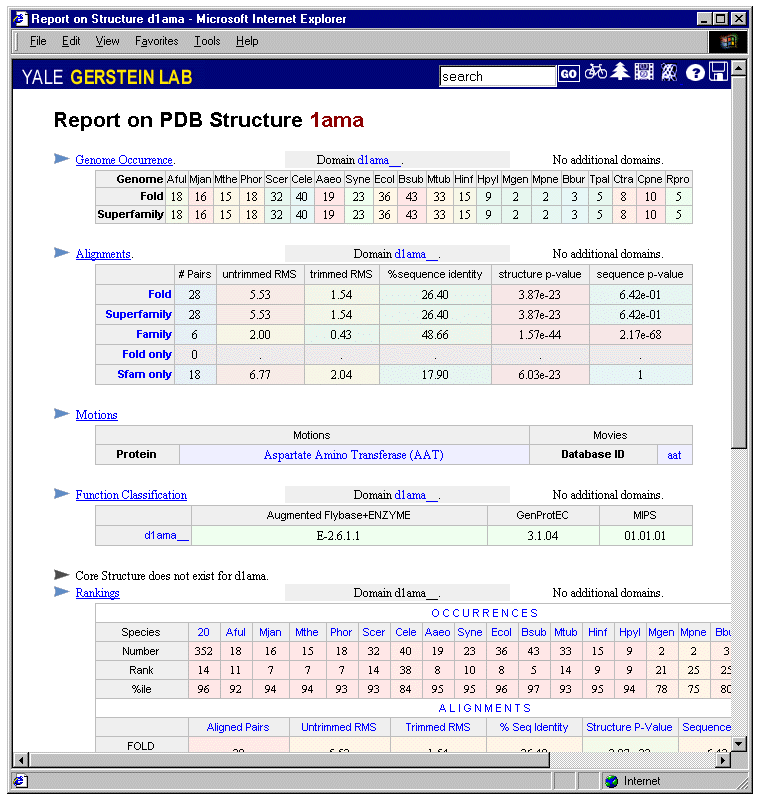
PDB report. This summarizes all the information concerning a domain or a representative PDB structure. It includes: (i) a summary of the occurrence report; (ii) a summary of the alignments available for structures in the same superfamily and fold; (iii) a description motions and motion-movies associated with the structure in the Macromolecular Motions database (30,31); (iv) a summary of the merged functional classification; (v) a core structure if available (64); (vi) ranking tables of the queried structure in various datasets. Figure 4 shows an example PDB report for structure 1ama.
Fold report. This lists all the SCOP domains that are associated with the queried fold and provides information (similar to that in the PDB report) that is common to all -- i.e. genome occurrence, alignment report, and rankings.
Summary and Discussion
We developed a web-based system for dynamically ranking protein folds based on disparate attributes, including fold occurrence in various genomes, expression level, alignment statistics, protein-protein interactions, motion statistics, and transposon sensitivity. Three ranking tools are provided -- Comparer, Profiler, and Correlator -- which can help users to place one fold in context of all other ones. The uniform system of ranks employed by PartsList provides a good framework for comparing different experiments and gaining a broad perspective on the complexity of genomes.
We anticipate that PartsList will have a relatively stable number of entries (i.e. folds), while for each entry the attributes that describe it will increase over time. In the future as experiments yield new information, PartsList will include more and more attributes. In particular, we anticipate much new expression information will be incorporated. We also plan to develop a form to allow automatic submission of new ranking attributes and encourage people to submit any ranking information.
Acknowledgments:
We thank NIH and Keck Foundation for support.References:
1. Chothia, C. (1992) Nature, 357, 543-544.
2. Brenner, S. E., Hubbard, T., Murzin, A., Chothia, C. (1995) Nature, 378, 140.
3. Wolf, Y. I., Grishin, N.V., Koonin, E.V. (2000) J. Mol. Biol., 299, 897-905.
4. Consortium, T. C. e. S. (1998) Science, 282(5396), 2012-8.
5. Berman, H., M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N., Bourne, P.E. (2000) Nucleic Acids Res., 28, 235-242.
6. Laskowski, R. A., Hutchinson, E.G., Michie. A.D., Wallace, A.C., Jones, M.L., Thornton, J.M. (1997) Trends Biochem. Sci., 22, 488-490.
7. Wang, Y., Addess, K.J., Geer, L., Madej, T., Marchler-Bauer, A., Zimmernan, D., Bryant, S.H. (2000) Nucleic Acids Res., 28, 243-245.
8. Ball, C. A., Dolinski, K., Dwight, S.S., Harris, M.A., Issel-Tarver, L., Kasarskis, A., Scafe, C.R., Sherlock, G., Binkley, G., Jin, H., Kaloper, M., Orr, S.D., Schroeder, M., Weng, S., Zhu, Y., Botstein, D., Cherry, J.M. (2000) Nucleic Acids Res., 28, 77-80.
9. Frishman, D., Heumann, K., Lesk, A., Mewes, H. W. (1998) Bioinformatics, 14, 551-561.
10. FlyBase. (1999) Nucleic Acids Res., 27, 85-88.
11. Tatusov, R. L., Galperin, M.Y., Natale, D.A., Koonin, E.V. (2000) Nucleic Acids Res., 28, 33-36.
12. Aach, J., Rindone, W., Church, G.M. (2000) Genome Res., 10, 431-445.
13. Bader, G. D., Hogue, C.W. (2000) Bioinformatics, 16, 465-477.
14. Xenarios, I., Rice, D.W., Salwinski, L., Baron, M.K., Marcotte, E.M., Eisenberg, D. (2000) Nucleic Acids Res., 28, 289-291.
15. Benson, D. A., Karsch-Mizrachi, I., Lipman, D.J., Ostell, J., Rapp, B.A., Wheeler, D.L. (2000) Nucleic Acids Res., 28, 15-18.
16. Murzin, A. G., Brenner, S.E., Hubbard, T., Chothia, C. (1995) J. Mol. Biol., 247, 536-540.
17. Orengo, C. A., Michie, A.D., Jones, S., Jones, D.T., Swindells, M.B., Thornton, J.M. (1997) Structures, 5, 1093-1108.
18. Holm, L., Sander, C. (1996) Science, 273, 595-602.
19. Gibrat, J. F., Madej, T., Bryant, S.H. (1996) Curr. Opin. Struc. Biol., 6, 337-385.
20. Madej, T., Gibrat, J-F., Bryant, S.H. (1995) Proteins, 23(356-369).
21. Bateman, A., Birney, E., Durbin, R., Eddy, S.R., Finn, R.D., Sonnhammer, E.L.L. (1999) Nucleic Acids Res., 27, 260-262.
22. Henikoff, J. G., Greene, E.A., Pietrokovski, S., Henikoff, S. (2000) Nucleic Acids Res., 28, 228-230.
23. Schultz, J., Milpetz, F., Bork, P., and Ponting, C.P. (1998) Proc. Natl. Acad. Sci. USA, 95, 5857-5864.
24. Hegyi, H., Lin, J., Gerstein, M. (2000) submitted.
25. Gerstein, M. (1998) Proteins, 33, 518-534.
26. Gerstein, M., Levitt, M. (1997) Proc. Natl. Acad. Sci. USA, 94, 11911-11916.
27. Jansen, R., Gerstein, M. (2000) Nucleic Acids Res., 28, 1481-1488.
28. Drawid, A., Jansen, R., Gerstein, M. (2000) Trends Genet., 16, 426-429.
29. Wilson, C. A., Kreychman, J., Gerstein, M. (2000) J. Mol. Biol., 297, 233-249.
30. Gerstein, M. and Krebs, W. (1998) Nucleic Acids Res., 26, 4280-4290.
31. Krebs, W., Gerstein, M. (2000) Nucleic Acids Res., 28, 1665-1675.
32. Park, J., Karplus, K., Barrett, C., Hughey, R., Haussler, D., Hubbard, T., Chothia, C. (1998) J. Mol. Biol., 284, 1201-1210.
33. Teichmann, S., Chothia, C., Church, G., Park, J. (2000) Bioinformatics, 16, 117-124.
34. Ross-Macdonald, P. C., P.S.R., Roemer, T., Agarwal, S., Kumar, A., Jansen, R., Cheung, K., Sheehan, A., Symoniatis, D., Umansky, L., Heidtman, M., Nelson, F.K., Iwasaki, H., Hager, K., Gerstein, M., Miller, P., Roeder, G.S., Snyder, M. (1999) Nature, 402, 413-418.
35. Jelinsky, S. A., Samson, L.D. (1999) Proc. Natl. Acad. USA., 96, 1486-1491.
36. Holstege, F. C., Jennings, E.G., Wyrick, J.J., Lee, T.I., Hengartner, C. J., Green, M.R., Golub, T.R., Lander, E.S., and Young, R.A. (1998) Cell, 95, 717-728.
37. Velculescu, V. E., Zhang, L., Zhou, W., Vogelstein, J., Basrai, M.A., Bassett, D.E., Jr, Hieter, P., Vogelstein, B., Kinzler, K.W.,. (1997) Cell, 88, 243-251.
38. Roth, F. P., Hughes, J. D., Estep, P.W., Church, G. M. (1998) Nature Biotechnology, 16, 939-945.
39. Spellman, P. T., Sherlock, G., Zhang, M.Q., Iyer, V.R.,, Anders, K. Eisen, M.B., Brown, P.O., Botstein, D., Futcher, B. (1998) Mol Biol Cell, 9, 3273-3297.
40. DeRisi, J. L., Iyer, V.R., and Brown P.O. (1997) Science, 278, 680-686.
41. Chu, S., DeRisi, J., Eisen, M., Mulholland, J., Botstein, D., Brown, P.O., Herskowitz, I. (1998) Science, 282, 699-705.
42. Richmond, C. S., Glasner, J.D., Mau, R., Jin, H., Blattner, F.R. (1999) Nucleic Acids Res., 27, 3821-3835.
43. Wixon, J., Blaxter, M., Hope, I., Barstead, R., Kim, S. (2000) Yeast, 17, 37-42.
44. Brenner, S. E., Koehl, P., Levitt, M. (2000) Nucleic Acids Res., 28, 254-256.
45. Uetz, P., Giot, L., Cagney, G., Mansfield, T.A., Judson, R.S., Knight, J.R., Lockshon, D., Narayan, V., Srinivasan, M., Pochart, P., Qureshi-Emili, A., Li, Y., Godwin, B., Conover, D., Kalbfleisch, T., Vijayadamodar, G., Yang, M., Johnston, M., Fields, S., Rothberg, J.M. (2000) Nature, 403, 623-627.
46. Ito, T., Tashiro, K., Muta, S., Ozawa, R., Chiba, T., Nishizawa, M., Yamamoto, K., Kuhara, S., Sakaki, Y. (2000) Proc. Natl. Acad. Sci. USA, 97, 1143-1147.
47. Harrison, P., Echols, N., Gerstein, M. (2000) submitted.
48. Hegyi, H., Gerstein, M. (1999) J. Mol. Biol., 228, 147-164.
49. Lipman, D. J., Pearson, W.R. (1985) Science, 227, 1435-1441.
50. Altschul, S. F., Koonin, E.V. (1998) Trends Biochem. Sci., 23, 444-447.
51. Brenner, S., Chothia, C. and Hubbard, T. (1998) Proc. Natl. Acad. Sci. USA, 95, 6073-6078.
52. Teichmann, S., Chothia, C., Gerstein, M. (1999) Curr. Opin. Struc. Biol., 9, 390-399.
53. Gerstein, M., Lin, J., Hegyi, H. (2000) Pacific Symposium on Biocomputing, 5, 30-42.
54. Lin, J., Gerstein, M. (2000) Genome Res., 10, 808-818.
55. Levitt, M. and Gerstein, M. (1998) Proceedings of the National Academy of Sciences USA, 95, 5913-5920.
56. Gerstein, M. and Levitt, M. (1998) Protein Science, 7, 445-456.
57. Gerstein, M. (1998) Folding & Design, 3, 497-512.
58. Brown, P. O. and Botstein, D. (1999) Nat Genet, 21(1 Suppl), 33-7.
59. Lipshutz, R. J., Fodor, S. P., Gingeras, T. R. and Lockhart, D. J. (1999) Nat Genet, 21(1 Suppl), 20-4.
60. Gerstein, M., Jansen, R. (2000) Curr. Opin. Struc. Biol.
61. Park, J., Lappe, M., Teichmann, S.A. (2000) submitted.
62. Bairoch, A. (1993) Nucleic Acids Res., 21, 3155-3156.
63. Riley, M., Labedan, B. (1996) In Neidhardt, F., Curtiss, III, R., Lin, E.C.C., Ingraham, J., Low, K.B., Magasanik, B., Reznikoff, W., Riley, M., Schaechter, M., Umbarger, H.E. (ed.), Escherichia coli and Salmonella: Cellular and Molecular Biology. ASM Press, Washington D.C., pp. 2118-2202.
64. Schmidt, R. B., Gerstein, M., Altman, R.B. (1997) Protein Science, 6, 246-248.
Figure and Table captions
Table 1
This table shows all the attributes ranked by PartsList. The formalism for specifying an attribute has two parts: an overall category, denoted by a single uppercase symbol, and some parameter choices, which are denoted by lower-case arguments to the first symbol. Some examples for folds will suffice to make this clear: G(aful) is genome occurrence of a particular fold in A. fulgidus; M(nhinges,goldstd) is the maximum value of the number of hinges statistic from surveying a set of motions in the gold-standard subset of the Macromolecular Motions Database, where this statistic is only calculated for the entries in the motions database that are associated with a particular fold; And J(pdball,inter) is the number of distinct types of protein-protein interactions found in a survey of the PDB subject to the restriction that the interactions must be between folds on different chains.
Figure 1
The overall structure of PartsList. Three tools (Profiler, Comparer, and Correlator) provide an easy way to access and manipulate the display of the dataset. With these tools, users can isolate interesting folds and obtain fold reports about them. Further clicks takes one to PDB report, which gives detailed information about an individual structural domain, including its genome occurrence, alignment information, molecular motions, functional annotation, interactions, and core structure.
Figure 2
Sample displays. (A) an sample Comparer display: the four selected attributes are genome occurrence in yeast, fold occurrence in E. coli, fluctuation of expression level for CDC28 synchronized yeast cell during the cell cycle, and the corresponding values for E. coli to heat shock. The folds are ranked in terms of fold occurrence in E. coli and the most common fold here is the TIM-barrel (represented by the SCOP domain d1aj2__). If one clicks the ?Display ranks? button, the values in the cells will be replaced by the ranks in their respective column. By clicking the ?re-rank? arrows, one can also obtain other views by sorting on other attributes. (B) Shows the occurrences of folds in 20 genomes in Profiler. (C) Shows the correlation between the fold occurrences in the A. fulgidus and S. cerevisiae (Aful and Scer) genomes. Both linear and rank correlation coefficients are calculated. The linear correlation coefficient is defined as: , where and are two vectors with N elements. Each element of the X vector is normalized thus: , where and are the average and standard deviation of the values of the original data vector X?, respectively. Y is normalized in a similar fashion. For two perfectly correlated datasets , while for two completely uncorrelated datasets . If we replace by its rank among all the other in the sample (i.e., 1,2,3?,N), then we get the rank correlation coefficient. A scatter plot is also shown to help in visualizing this correlation.
Figure 3
The relation between the number of functions associated with a protein fold and the number of distinct protein-protein interactions it has (based on a survey of the PDB databank). This relationship can be displayed both in Comparer (left) and Correlator (right).
Figure 4
An example PDB report for structure 1ama. The report summarizes the relevant information for this domain, including genome occurrences, alignment, motions, function classification, core structure and rankings. By clicking on the headers, one can get the detailed reports for these quantities.
{kind=link}
{kind=link}
{kind=link}
{kind=link}