Mark Gerstein's Research Program and Coupled List of Papers (02.01.28)
Research Program,
Figures page 1 and 2
(ppt of figures),
Coupled Papers List
Numbered consistently and 12 best papers flagged with * and best review by #
(Providing Coupled Annotation to the List of Publications
**)
Computational Proteomics:
Genome-scale Analysis of the Structure, Function and Evolution of
Proteins
As we move into the new century, the human genome and those of many other
organisms, comprising billions of basepairs, have been sequenced. The number
of known structures of protein domains, which provide the primary way to
interpret gene sequences as functioning molecules, has risen to more than
25,000. And a variety of functional genomics experiments, such as cDNA
microarrays, are providing large amounts of standardized data on each gene
in the genome. Remarkably, with the simultaneous advance of computer
technology, all this information fits easily onto a laptop. Interpreting it,
however, will require new approaches.
Broadly, the goal of my laboratory is to make sense of this data deluge, by
carrying out integrative surveys and systematic data mining on the expanding
amount of biological information. Specifically, we are focused on computational
proteomics: understanding the structure, function, and evolution of proteins
through analyzing populations of them in the databases and in whole-genome
experiments. Through such work we believe we can address two central post-
genomic challenges: understanding genes in detail and interpreting the regions
between genes. With regard to the first challenge, we are trying to predict
protein function on a genomic scale and trying to understand how we get a range
of structural and functional diversity from a limited repertoire of protein folds.
With regard to the second challenge, we are analyzing protein fossils
(pseudogenes) in intergenic regions.
The ongoing research program in the lab extends and expands previous work as
described below, with work broadly falling into three areas: (i) analysis of
structures, focused on understanding protein motions in terms of packing; (ii)
analysis of sequences, focused on surveying the occurrence of folds and families
in genomes (both for genes and pseudogenes); (iii) analysis of functional
genomics data, focused on trying to predict protein function on a genomic scale.
Our work is fundamentally data-driven and different in conception from previous
computational work related to proteins, which often concentrated on describing
the physical process of folding or predicting 3D structure given an amino-acid
sequence. It is closely coupled with experiment and involves a number of detailed
collaborations with experimentalists. Finally, our work, which falls into the new
discipline of bioinformatics, is fundamentally interdisciplinary in character,
combining questions drawn from biology with quantitative approaches from computer
science and physics.
A. Analyzing Structures: Quantifying the Diversity in a Limited Number of Folds
1. Structure Comparison of Protein Folds
A starting point for our analysis is the concept of a finite list of biological
parts. One of the most direct ways of appreciating this can be illustrated using
protein folds: It is believed that there is a large but limited number of folds
(estimated to be ~5000), and a library of them represents a most important resource
for biology. To build a library of folds, one needs some statistical or heuristic
definition of what a fold is, a way of clustering together all the structures with
a given fold, and intelligent techniques for matching up sequences with unknown
structure to those with known structure. We are working on a number of these topics
[29,35,26] - on the one hand, trying to build up our own classifications of
structures and, on the other hand, simultaneously trying to integrate structural
classifications developed by others (such as scop and cath) into our own analyses.
In particular, we have developed a way to use existing structural classifications
as scaffolds for integrating diverse genomic information [72*,
PartsList.org, Fig. 1].
2. Classification of Protein Structural Flexibility in a Web Database
An important aspect of a fold library is its use in comprehensively surveying
protein flexibility and conformational variability -- measuring how much each
part in the master parts list can vary in shape. We are classifying all
instances of conformational variability into a web-accessible database
[38*, 99, MolMovDB.org]. Part of this project involves devising a
system for characterizing protein motions in a highly standardized fashion,
in terms of a few key statistics, such as the location of hinges and the
degree of rotation about them. We have developed a web server that, given
two coordinate sets, automatically does this
(producing "morph movies" as a by-product) [57, Fig. 2]. Our classification of
motions is based on the packing at internal interfaces [52]. Motions are
identified as shear or hinge, based on whether or not a well-packed interface
is maintained between the mobile elements throughout the motion.
3. Geometric Measurement of Packing and its use in Rationalizing Protein Motions
Our motions classification scheme is motivated by the fact that protein interiors
are packed exceedingly tightly, and the tight packing at internal interfaces
greatly constrains the way proteins can move. Our past research has involved
measuring the packing efficiency at different interfaces (e.g. interdomain,
protein surface) using specialized geometric constructions (e.g. Voronoi polyhedra),
in conjunction with limited amounts of molecular simulation [30,33,40,81,84, Fig. 3].
We recently developed a new parameter set for these calculations, which includes
self-consistent VDW radii and standard volumes for each atom type
[48*,83,107].
B. Analyzing Sequences: Surveying the Occurrence of Proteins in Genomes
1. Computational Structural Genomics: Comparing Folds between Proteomes
As more genomes are sequenced, and structures, determined, it has become
increasingly possible to characterize a substantial fraction of the folds used
in a given organism -- statistically, in the sense of a population census.
This allows us to see whether particular folds are more common in certain
organisms than in others. We were the first laboratory to address questions of
this sort, performing comparisons of genomes in terms of folds[34*].
In these and other surveys we have found that a number of
folds, such as TIM-barrels, occur in every (analyzed) genome, while other folds are
missing from certain genomes [32,43,47,60,104]. Fold occurrence can be used
to build whole-genome trees, with the distances between organisms defined in terms
of the presence or absence of specific folds in the whole genome[41,58*,
GeneCensus.org, Fig. 4]. This contrasts to traditional phylogenies, which group
organisms based on sequence similarity of individual genes.
While we found that the specific most common folds often differed between genomes,
in all cases the occurrence of folds (and many other aspects of genomic biology)
tends to follow power-law statistics, with a few common ones and many rare ones.
We have proposed a simple evolutionary model that naturally gives rise to these
statistics [92, Fig. 5]. Finally, we found that
there were many global, statistical differences between folds from different
phylogenetic groups -- e.g. with longer and more numerous all-beta proteins from
eukaryotes than prokaryotes and more salt-bridge pairs in thermophiles [42,65,87].
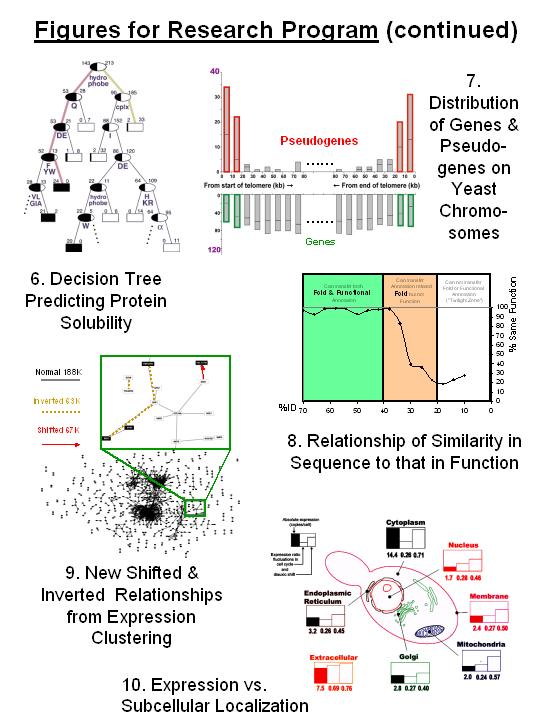
2. Experimental Structural Genomics: Target selection & Data mining
Our surveys on folds in genomes are coupled to collaborations with
crystallographers and NMR spectroscopists carrying out experimental
structural genomics, trying to determine structure in high-throughput fashion.
In particular, we have done target selection, database design, and datamining
for one of the structural genomics centers, and this has enabled us to develop
systematic rules to predict expressibility and solubility of proteins
[61,63,69,76*, Fig. 6, NESG.org].
3. Surveying Pseudogenes in Intergenic Regions, Looking for Ancient Protein Relics
In addition to analyzing the occurrence of folds and families within the "living"
proteome, we can also use them to survey the "dead" pseudogenes and pseudogeneic
fragments in intergenic regions. We were one of the first to perform comprehensive
surveys of pseudogenes on a genome-wide scale in terms of protein families, which
we did for the worm[73*]. We have done subsequent surveys
of yeast, fly and human [97,98,103, Fig. 7]. Collectively, these allow us to
determine the common "pseudofolds" and "pseudofamilies" in various genomes and to
address important evolutionary questions about the type of proteins that were
present in the past history of an organism. In particular, we have found that
duplicated pseudogenes tend to have a very different distribution than one would
expect if they were randomly derived from the population of genes in the genome.
They tend to lie on the end of chromosomes and have an intermediate composition
between that of genes and intergenic DNA. Most importantly, pseudogenes tend to have
environmental-response functions. This may be related to their being resurrectable
protein parts, and we propose a potential mechanism for achieving this in yeast
[103*]. Processed pseudogenes, which are common in the human
genome, have a very different character. They appear to be randomly inserted from mRNA
pool and, hence, show an obvious relationship to mRNA level and intergenic region size.
Our pseudogene work forms a valuable backdrop to experimental work aimed at accurately
identifying genes and annotating genomes as well as probing the sequence
characteristics of intergenic regions [94,101].
C. Predicting Protein Function on a Genome Scale, through Data Integration
Because of its size and complexity, individual experimentation for functional
annotation of every gene in the human genome is not possible. Thus, a central
problem in proteomics is how to determine protein function on a large-scale.
There is a wide range of computational approaches to this problem, from
traditional sequence pattern matching to newer approaches that deal with
interpreting microarray data. As outlined below, we are pursuing all of these.
1. Functional and Structural Annotation Transfer between Similar Sequences
One of the most used (and abused) techniques in genome analysis is "annotation
transfer", carrying over information related to a variety of properties (e.g.
structure and function) from a known sequence in the databases to an unknown
one in the genome that is similar to it. We are using manually built
classifications of protein folds and functions to provide benchmarks to measure
to what degree structural and functional annotation can be reliably transferred
between similar sequences, particularly when similarity is expressed in modern
probabilistic language [31,39,64]. The key issue here is defining appropriate
sequence similarity thresholds for the transfer of functional annotation, and
based on our analysis, we have been able to find clear thresholds (e.g. 40%
identity) for single and multi-domain proteins [55*, 89, Fig. 8].
2. Predicting Function from Structure? Fold-Function Relationships
Another method to obtain the function of an uncharacterized protein is through
determining its 3D structure and then looking for structural similarities to
proteins of known function. This is a central idea in both structural genomics
and structure prediction. To address this issue, we have measured, globally,
the degree to which fold is associated with function[45*,70].
We find there is a relationship but it is not strongly
predictive. There are also interesting correlations; for instance, the
association of enzymatic functions with alpha/beta folds appears to be fairly
universal but not present in particularly ancient proteins.
3. Clustering Microarrray Data and Relating it to Protein Features
A new approach for getting at protein function is clustering gene-expression
data from microarrays -- genes that cluster together may be functionally
related. We have been performing many such analyses focusing on cross-
referencing expression clusters to broad "proteomic categories," such as
functions and families [54,77].
We have found this approach averages away much of the noise in
expression data. We have also worked extensively on relating mRNA expression
to protein abundance in terms of broad categories [78,105]. In terms of
function prediction, we have developed
a new method of clustering expression data that finds many time-shifted and
inverted relationships in addition to the simultaneous relationships found in
other studies, and we have developed a way of quantifying how much a given
expression clustering predicts protein functional role or protein-protein
interactions[66,91*,95, Fig. 9]. Overall, we find that while expression
clustering identifies many new and suggestive functional relationships, it is
not strongly predictive in a global sense.
4. Integrating Diverse Information to Predict Function, including the Literature
In addition to microarrays, many other types of functional genomics experiments
have recently appeared. We have been involved with a number of these --
highlights include measuring the effect of genome-wide transposon insertions
and using proteome chips to assay protein binding comprehensively [53,68,86].
We find that cluster
analysis of these experiments correlates better with biochemical and phenotypic
function than does expression clustering. However, no individual experiment
provides a full description of function. Integrating many experiments together
with "traditional" sequence information (e.g. motifs, composition, and database
matches) clearly should (and indeed does) give better functional predictions,
and we believe one the most important uses for proteins and protein families
is as scaffolds for achieving large-scale integration[71,79,82,93*].
One often overlooked type of information that is critical to integrate into
biological databases is journal articles, which contain the overwhelming bulk
of what is known about function, albeit in unstructured form. Connecting
databases and journals is a vast challenge, and in this area we have identified
issues, advocated approaches and developed standards [44,46,49,50,59,74,88,90,102].
We envision a future, where
there will be less distinction between databases and journals. One will be able
to both find understandable prose in database entries and do computation directly
on specially constructed parts of journal articles. Such a scenario will help
overcome many of the problems now facing biological databases, including quality
control, attribution of credit, and error correction.
5. A Practical Example of Data Integration, Predicting Subcellular Localization
Although easy to advocate, data integration is tricky in practice, as it often
involves giving highly heterogeneous features -- such as expression timecourses,
two-hybrid pairs, and sequence motifs -- different weights within a single
mathematical formalism. In one particular context, we have been able to
successfully integrate many features for function prediction: predicting
subcellular localization[62*,67,85,106, Fig. 10] We found that the
localization of a protein is related to the expression level of its associated
gene -- e.g. lowly expressed proteins were more likely to be destined for the
nucleus than cytoplasm. We then used a Bayesian system to seamlessly integrate
this expression observation with traditional sequence motifs and essentiality
information and predict localization for all the proteins in yeast.
Summary & Future Directions
In summary, our lab was one of the first to work on comparing genomes in
terms of folds. We were also one of the first to do genome-wide surveys of
pseuduogenes and perform integrated data mining on functional genomics data.
Our tools for analyzing motions and packing are well used, and our scoring
schemes for annotation transfer are practically useful in genomics. In general,
we believe our combination of comparative genomics with traditional biophysical
and structural calculations gives us a broad and unique perspective on questions
in bioinformatics.
There are number of future directions that we are pursuing. These are all direct
extensions of our current program. In terms of structure, we would like to develop
"rich templates" for the most common folds (e.g. helical bundles and TIM-barrels)
that highlight their flexibility about around a common framework. We envision that
these will be useful in protein design, in simplifying complex conformational
changes, and in providing annotation to the newly solved structures that do not
have novel folds (e.g. from structural genomics). In terms of genome analysis, we
plan to scale up in two ways. On the one hand, we would like to enlarge our surveys
of pseudogenes to the entire human genome, and, on the other, we would like to focus
on comparing the human proteome against those of pathogens, trying to identify
unique pathogen proteins as antibiotic targets. Of particular interest here are
membrane proteins, which are often exposed on the surface. In terms of function, we
plan to extend our Bayesian system for localization to the prediction of protein-
protein interactions. We believe that integrating the new data from protein-chip
experiments and from many of the many recent protein interaction sets creates for
the first time a large enough database to be able to rigorously tackle this problem.
** NOTE: This document is closely coupled to my publication list in
the following fashion: Almost each publication between 1/97 and early 2002 is
referred to once and only once here, using its bibliography number. My top
publications and reviews are highlighted, and the sentence before the citation,
which explains their significance, is italicized. Thus, from glancing at the
highlighting, one should be able to get a quick bullet-point summary of the main
papers. To keep things simple, no attempt has been made to refer to the
scientific literature generally, and this document should not be construed as a
review of the field.
{kind=link}
{kind=link}